Similar interests
- Non Gamstop Casinos
- Best Non Gamstop Casinos
- Non Gamstop Casinos
- Non Gamstop Casinos
- Casinos Not On Gamstop
- Gambling Sites Not On Gamstop
- Melhores Casinos Online
- Crypto Casino
- Non Gamstop Casinos UK
- Non Gamstop Betting Sites
- Online Casino
- Slots Not On Gamstop
- UK Casinos Not On Gamstop
- Casinos Not On Gamstop
- Casinos Not On Gamstop
- Casino Online Non Aams
- Non Gamstop Casinos UK
- Meilleur Casino En Ligne Français
- Gambling Sites Not On Gamstop
- Non Gamstop Casino Sites UK
- Non Gamstop Casino
- Uk Sports Betting Sites Not On Gamstop
- Siti Casino Online Non Aams
- Best Non Gamstop Casino
- Casino Non Aams
- Non Gamstop Casino UK
- Migliori App Casino Online
- Casino En Ligne Belgique Bonus
- Site Paris Sportif Tennis
- オンカジ スロット おすすめ
- 本人確認不要 カジノ
- крипто казино онлайн
- 카지노게임사이트
- Meilleur Casino En Ligne France
- Meilleur Casino En Ligne 2026
- Casino Italiani Non Aams
- Meilleur Casino En Ligne
- Nouveaux Casino En Ligne
- Crypto Casinos Malaysia
- Casino Machine A Sous
- Casino En Ligne Francais
- ポーカー オンライン
- Casinos En Ligne
Human Genome Variation
|
Each individual has two copies of the human genome: one maternal, one paternal. Any two human genomes differ in about 3M single nucleotide variants, 1/10 as many small insertions and deletions, and thousands of larger structural variants (SVs). Human genome variation is important:
We develop algorithms for modeling and discovering human genome variation, and for leveraging variation to study ancestry, relatedness and phenotype. Examples of our recent work include:
|
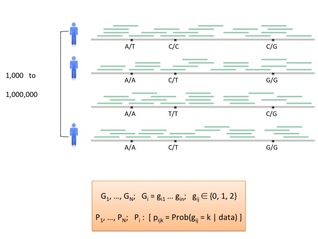
Sequencing and genotyping a large population.
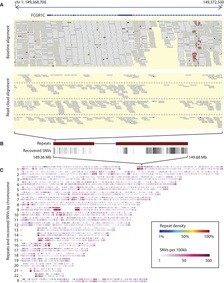
Uncovering dark variants in the human genome.
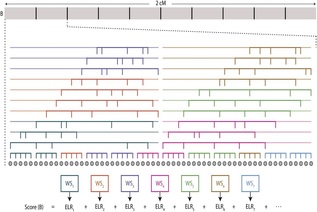
Modeling LD and inferring relatedness.
|