Similar interests
- Non Gamstop Casinos
- Best Non Gamstop Casinos
- Non Gamstop Casinos
- Non Gamstop Casinos
- Casinos Not On Gamstop
- Gambling Sites Not On Gamstop
- Melhores Casinos Online
- Crypto Casino
- Non Gamstop Casinos UK
- Non Gamstop Betting Sites
- Online Casino
- Slots Not On Gamstop
- UK Casinos Not On Gamstop
- Casinos Not On Gamstop
- Casinos Not On Gamstop
- Casino Online Non Aams
- Non Gamstop Casinos UK
- Meilleur Casino En Ligne Français
- Gambling Sites Not On Gamstop
- Non Gamstop Casino Sites UK
- Non Gamstop Casino
- Uk Sports Betting Sites Not On Gamstop
- Siti Casino Online Non Aams
- Best Non Gamstop Casino
- Casino Non Aams
- Non Gamstop Casino UK
- Migliori App Casino Online
- Casino En Ligne Belgique Bonus
- Site Paris Sportif Tennis
- オンカジ スロット おすすめ
- 本人確認不要 カジノ
- крипто казино онлайн
- 카지노게임사이트
- Meilleur Casino En Ligne France
- Meilleur Casino En Ligne 2026
- Casino Italiani Non Aams
- Meilleur Casino En Ligne
- Nouveaux Casino En Ligne
- Crypto Casinos Malaysia
- Casino Machine A Sous
- Casino En Ligne Francais
- ポーカー オンライン
- Casinos En Ligne
Read Cloud Technologies
|
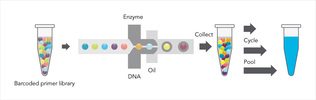
Today, the most cost effective technologies for sequencing a genome (e.g., Illumina and Complete Genomics) have a significant limitation: the sequence reads are short. For instance, reads produced by HiSeq today are up to 150bp in length. As a result, the following are challenges of short reads:
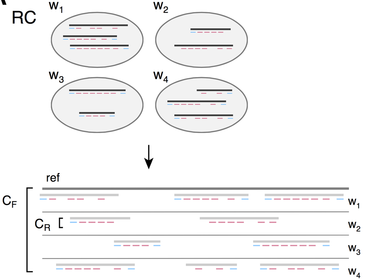
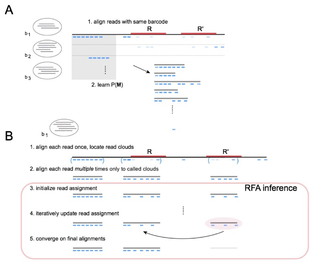
We develop algorithms for many of the above problems. We collaborate extensively with 10X to apply the latest technology to projects in cancer genomics, metagenomics, and human genome variation identification. One of our latest algorithms, RFA, employs a Markov random field (MRF) to model the process of sequence preparation with read clouds, and map reads to the entire genome including dark regions. As a result, variant calling on the previously dark 6% of the genome is now greatly facilitated. Fun reading: my favorite "unscored" NIH proposal. I thank Annelise Barron for her input. |
|